Today I officially ended a 10-year stint on the Scientific Advisory Board of the Frontotemporal Dementia Disorders Registry (FTDDR). It maintains a list of people with the “FTD disorders,” loosely defined to include PSP and corticobasal degeneration as well as the standard forms of FTD. The FTDDR is the only widely-available registry for PSP and collaborates closely with CurePSP. It is supported financially by the Rainwater Charitable Foundation, which provides generous funding and infrastructure support for research on tau-based disorders, especially PSP.
Registering with the FTDDR would allow you to contribute information on your condition to a powerful tool for researchers, philanthropists, government policymakers and patient support organizations. It could serve to introduce you to drug companies and academic researchers seeking patients for treatment trials or observational (i.e., non-treatment) research. Of course, you can choose which information about yourself to include and not to include, and you can opt out of consideration for treatment trials if you like.
The registry is designed to safeguard your confidentiality. When the registry personnel supply data to outside organizations , the registrants’ identifying and contact information is stripped away first. If an outside organization wishes to contact contact you about a research project, the registry office would manage that for them. In that way, you could decline without revealing your identity to the outside organization.
I highly recommend that you register. Just go to https://ftdregistry.org/ and follow the instructions.
Did you know that Toronto is the most ethnically diverse city in the world? Besides, it’s a pretty big place, with about 6 million people in its metropolitan area. Besides that, Toronto is home to one of the top PSP research institutions in the world, the Rossy Centre at the University of Toronto, and Canada has universal, free medical insurance, which removes financial impediments to diagnosis and treatment. That’s why Toronto is a great place to answer the question as to whether the prevalence of PSP is uniform across groups of different ethnic backgrounds.
Dr. Blas Couto and colleagues have done just that, reporting their results in the current issue of Parkinsonism and Related Disorders. They tabulated ethnicity for 135 patients with PSP living in the officially designated Toronto area and seen at their center as patients from 2019 to 2023. The group proved unusual in that only 4.4% had the PSP-Parkinsonism variant, compared with figures elsewhere of around 25% to 40%. The group with PSP-Richardson syndrome comprised 68% rather than the usual 45% to 55% and the other variants gave the expected percentages.
The ethnicities they considered were actually geographical areas, not exactly race, whatever that is. (“Race” has no scientific definition, anyway.) The categories were — and this is directly copied/pasted from the paper:
East and southeast Asia, including China and Pacific Islands such as Philippines
Southern Asia, including India, Pakistan and middle east countries
Africa
South America, Central America and Mexico
West Indies, including Guyana, Haiti, Bahamas, Lesser Antilles such as Barbados, Trinidad and Tobago, Dominica, Grenada, Saint Kitts, Antigua and Barbuda, Santa Lucia
Europe, Australia and North America, excluding Mexico.
The analysis compared the frequencies of these demographics to those from the census for people aged 65 and older living in the Toronto metro area.
The result was that the southern Asia group was moderately over-represented among the patients with PSP. That group accounted for 11.5% of the general population but 25.2% of the PSP population at the researchers’ center. That was statistically significant at the <.001 level, meaning that the chance of its being a false-positive fluke are less than 1 in 1,000.
They also compared the six groups to one another with regard to the PSP subtypes, finding the same southern Asia group to include more PSP-progressive gait freezing (17.7%) and PSP-corticobasal syndrome (14.7%) than the European-derived patients (6.4% and 9.5%, respectively).
Couto et al mention the possibility of some sort of genetic effect, but the literature offers no clues as to what that might be, and they cite three previous papers from the UK showing no difference in PSP prevalence between whites and southern Indians there. Could something in the food or water in Toronto affect Asians disportionately? The title of this post offers an unserious possibility, but you get the idea.
Chin-stroking on that aside, now is when the rest of us try to poke holes in the findings. Here are my efforts:
Do the ethnic percentages in their PSP practice or in medical institution as a whole accurately reflect those of the Toronto area? That would be easy to measure. I ask because in the US in recent years, the medical profession has acquired a disproportionate representation of people of southern Asian background. Could that group therefore trust academic physicians more and seek their care more readily than do other ethnic groups?
The patient mix of a highly specialized practice like that at the Rossy PSP Centre is subject to the referral habits of outside neurologists. Neurologists who feel less comfortable with the atypical Parkinsonisms may be more likely to refer patients. Perhaps that applies to a few neurologists practicing in heavily southern Asian neighborhoods in the Toronto area.
Despite the universal availability of free medical care in Canada, racially-based disparities in health and care access do exist there. Couto et all cite this article.
This is a univariate comparison, meaning that it didn’t correct for a hypothetical effect of other health issues that might be more common in the southern Asian population. Not that I know what those might be.
Although the analysis age-matched the PSP group with the general population by confining itself to the over-65 group, that may not have been enough. Perhaps working-age people immigrating from southern Asia brought elderly parents with them more often than did those immigrating from elsewhere, thereby skewing the over-65 group towards the 80s and 90s and increasing the measured prevalence of an age-related disease like PSP.
An intriguing finding. Hopefully this paper will stimulate others to dig deeper. That would be a victory for any scientific paper.
I have some homework for you all: Make sure that the diagnosis of PSP has been accurately coded in the doctor’s records.
Two researchers in Scotland, Diane Swallow and Carl Counsell, published a paper this week reporting that every single patient carrying the code for PSP (G23.1) did in fact have PSP on evaluation of their detailed records. However, only 52% of the patients whose records showed clear PSP had that code recorded in their records. In 45% of individuals with PSP, an incorrect code of G12.2 was recorded. That’s the code for a condition called “progressive bulbar palsy,” which is a form of amyotrophic lateral sclerosis (ALS; Lou Gehrig disease) that affects the muscles of the face, mouth and throat disproportionately. (The “bulb” is another term for the medulla, the part of the brain from which most of the nerves to those structures arise. It does look sort of like a light bulb or tulip bulb.) PGP is much more rare than PSP and does not include most of its features, but the reason for the confusion is obvious.
Why is this important? Because accurate statistics on PSP’s prevalence are used by for scientific researchers, medical care planners, government regulatory agencies, insurance companies, pharmaceutical companies, granting agencies, and philanthropists.
So, next chance you have, ask the neurologist’s office clerk if the diagnosis of PSP rather than progressive bulbar palsy is listed in the medical record’s formal problem list and if the code assigned to it is in fact G23.1. If it’s not, and if the clerk tells you that they don’t have the access necessary to fix it, make sure that they pass the information along to whoever does have that access, and then it’s your job follow up to make sure it’s been taken care of.
In response to a reader’s request, here’s a brief description of the mechanism of action of Relyvrio, which is a combination of two drugs, sodium phenylbutyrate and taurursodiol. The text in bold italics below is copied verbatim from the supplementary material attached to the publication reporting the results of the first ALS trial. The same explanation applies to PSP and other neurodegenerative diseases. You may feel that any treatment that claims to address all of those complex diseases is claiming too much, and you could be right. But stranger things have happened. If you want more scientific detail, see the five references below. Note that Reference 5 discusses release of cytochrome C from mitochondria. That’s a cell signalling compound that causes cells to start up their “suicide machine,” more formally called the apoptotic pathway. Cells undergo apoptosis when they’re not working well or as a normal “pruning” procedure during growth and development. Taurursodiol prevents that from happening as easily.
Endoplasmic reticulum stress or dysfunction associated with protein misfolding and aggregation has been implicated in the pathogenesis of ALS,[1] as has disruption of mitochondrial function and structure.[2] Sodium phenylbutyrate is a histone deacetylase inhibitor that has been shown to upregulate heat shock proteins and act as a small molecular chaperone, thereby ameliorating toxicity from endoplasmic reticulum stress.[3,4] Taurursodiol recovers mitochondrial bioenergetic deficits through several mechanisms, including by preventing translocation of the Bax protein into the mitochondrial membrane, thus reducing mitochondrial permeability and increasing the apoptotic threshold of the cell.[5]
1. Jaronen M, Goldsteins G, Koistinaho J. ER stress and unfolded protein response in amyotrophic lateral sclerosis—a controversial role of protein disulphide isomerase. Front Cell Neurosci 2014;8:402.
2. Mehta AR, Walters R, Waldron FM, et al. Targeting mitochondrial dysfunction in amyotrophic lateral sclerosis: A systematic review and meta-analysis. Brain Commun 2019;1:fcz009.
3. Kaur B, Bhat A, Chakraborty R, et al. Proteomic profile of 4-PBA treated human neuronal cells during ER stress. Mol Omics 2018;14:53-63.
4. Suaud L, Miller K, Panichelli AE, Randell RL, Marando CM, Rubenstein RC. 4-Phenylbutyrate stimulates Hsp70 expression through the Elp2 component of elongator and STAT-3 in cystic fibrosis epithelial cells. J Biol Chem 2011;286:45083-92.
5. Rodrigues CM, Solá S, Sharpe JC, Moura JJ, Steer CJ. Tauroursodeoxycholic acid prevents Bax- induced membrane perturbation and cytochrome C release in isolated mitochondria. Biochemistry 2003;42:3070-80.
As explained in my post of March 10 (“No panacea”), the trial drug Relyvrio was found not to help amyotrophic lateral sclerosis. This despite positive results last year from a smaller trial that had prompted the FDA to provisionally approve the drug for ALS. After this new result, the company, Amylyx, withdrew the drug from the market. However, an ongoing trial of Relyvrio for PSP will continue.
Relyvrio is also being tested not only in PSP, but also in patients with Wolfram syndrome. In case you haven’t heard of this genetic condition that’s about 2% as common as PSP, it causes a combination of insulin-requiring diabetes, excessive urine production (“diabetes insipidus”), blindness from degeneration of the optic nerve, and deafness, along with neurological issues such as seizures, mild cognitive loss and loss of respiratory drive. Wolfram syndrome starts at an average age of only six, with a range of six weeks to 19 years. Sufferers die by age 40, usually from complications of diabetes or from respiratory failure. A sad picture, indeed.
So what’s the encouraging news? Amylyx issued a press release on April 10 (three days ago) reporting interim results halfway through their 48-week, 12-patient, unblinded trial of Relyvrio in adults with Wolfram syndrome. Eight of the 12 patients had completed the trial by that date. The levels of a protein involved in the synthesis of insulin, called C-peptide increased and do so more promptly after a meal. C-peptide is a standard test in medical practice to assess the severity of diabetes. There was also a small improvement or lack of worsening in hemoglobin A1C; the fraction of the day in the normal blood glucose range; visual acuity as measured by a standard wall chart; and global impressions by the doctor and patient (separately) of how well things are going overall. Over 24 weeks, these patients would have been expected to worsen, on average, not to improve or stabilize as these eight patients apparently did.
Keep in mind that the comparator wasn’t a placebo group, but “historical controls,” meaning patients from previous research or from the study doctors’ regular practice. (Unfortunately, the company’s press release didn’t say just how much people with with Wolfram syndrome would be expected worsen in these measures over 24 weeks.) This opens the possibility that the patients in the study might simply have taken better care of themselves, knowing they were being tested in the trial. Another possibility is that the doctors themselves fell victim to their hopes for the patients, providing more aggressive general management of their symptoms during the trial. That’s why we do double-blind trials. If the current trial gives favorable results after all 12 patients have completed the 48 weeks, then presumably Amylyx will move to a larger and double-blind trial.
This is, provisionally, slightly good news for people with Wolfram syndrome, but what about PSP? Both diseases involve abnormalities in the “unfolded protein response” (UFR) in the brain cells’ endoplasmic reticulum (ER). After amino acids are strung together in the cell’s nucleus to make proteins, they’re transported to the ER, where they’re folded into the patterns they need to do their jobs. In both Wolfram syndrome and PSP, there’s an abnormal overactivity of the UFR, and Relyvrio inhibits it.
To date, we know of 11 genes that each confers a slightly increased risk of developing PSP. The most important is the MAPT gene, which encodes the tau protein. The next-most important is a gene called EIF2AK3, which encodes a protein called PERK, which is an important part of the unfolded protein response.
So let’s await the final results in this early-phase trial of Relyvrio in Wolfram syndrome. More to the point, let’s await results from the 600-participant, double-blind trial of Relyvrio in PSP, which has only just started recruiting and should end in mid-2027.
Disclosure: I’m a paid consultant for Amylyx. I assisted in the design of their PSP trial and in teaching the study doctors how to properly use the PSP Rating Scale. I have no stock in the company or any other financial interest in their commercial success.
You’ve probably heard about the disease called frontotemporal dementia (FTD). It produces many of the same cognitive and behavioral symptoms as PSP, but more intensely, with much less of PSP’s other symptoms. Familial forms of FTD are much more common than in PSP, amounting to about 20% of all cases. Of those, about a quarter have a mutation in their MAPT gene – that’s the one that encodes the tau protein. (Most of the rest have mutations in two obscure genes called “C9orf72” and “progranulin.”) As for nearly all neurodegenerative diseases, a protein aggregates in the affected brain cells in FTD and that protein corresponds to the specific mutated gene in that individual. So, MAPT-associated genetic FTD is a tauopathy very much like PSP, but in a slightly different set of brain cells.
Neurologists at UCSF and the Mayo Clinic are leading a large, multi-institutional observational study of FTD called “ALL-FTD.” They’re gathering histories, neurological exams, imaging, skin biopsies, blood, and most relevant here, spinal fluid. They re-gather much of this every year and track the patients’ progression. The idea is to find diagnostic markers not only to diagnose the disease, but to predict its onset in healthy mutation carriers and to predict the progression of the disease in those who already have symptoms. All of this can be useful in designing clinical treatment trials and in patient and family counseling.
Using spinal fluid from 116 people with FTD mutations and 39 controls, the ALL-FTD neurologists analyzed the levels of over 4,000 proteins. They found some of the proteins increased or decreased as functional modules. That means that subgroups of the affected proteins tended to share a common function in the brain. Then they tested spinal fluid from people with ordinary, non-familial PSP-Richardson syndrome for the same protein disturbances, and found some. In fact, all 31 of the functional modules disrupted in genetic FTD were disrupted in PSP as well. (This research article is posted on a site for manuscripts not yet peer-reviewed called “Research Square.”)
Mind you, this doesn’t mean that non-familial PSP is actually caused by genetic mutations in the three FTD-related genes. We still don’t know the initial cause of PSP. But the study does show that familial FTD and non-familial PSP share some very fundamental similarities.
The most important modules disrupted in the two conditions relate to the brain cells’ “extracellular matrix.” That’s the soup of chemicals outside and between the brain cells that serves many protective and nutritive functions. I suspect that it descended from the coat of slime secreted by our ocean-dwelling, single-celled ancestors. In ourselves it functions in cell growth, fetal development and injury repair as well as providing physical protection against trauma and a trap for nutrients floating by. It’s easy to see how a genetic or non-genetic defect in the contents our brain’s extracellular matrix could be a problem.
So, let’s add the extracellular matrix to our list of potential drug targets in PSP.
In response to a reader’s question, here’s the most current list I can create of current or imminent clinical pharmacological neuroprotective treatment trials in PSP. Maybe soon I’ll list the trials not covered by that list of adjectives, but you can do that yourself at www.clinicaltrials.gov.
For more information, including a phone number or email address, go to www.clinicaltrials.gov and search on the National Clinical Trials number shown or on the disease and drug names. For the trial in Australia/New Zealand, use the link given.
Relyvrio (AMX-0035): A orally-administered combination of two existing medications to help brain cells resist the PSP process. Will recruit 600 patients in the US, Canada, Europe and Japan. NCT06122662
NIO-752: An antisense oligonucleotide given by spinal injection once a month. Still recruiting only in Germany and the UK. A small, 3-month trial primarily designed to assess safety and tolerability. NCT04539041
Sodium selenate: A year-long trial of an oral drug. Australia only. ACTRN12620001254987
GV-1001: a subcutaneously protein fragment administered daily by subcutaneous injection. A small trial primarily designed to assess safety and tolerability. South Korea only. NCT05819658
Set to start in a few months is the study of FNP-223. This year-long trial will take place at dozens of sites in the US, Europe and Japan. It’s an oral pill taken 3 times a day that reduces the abnormal phosphorylation and misfolding of tau. It’s not yet on www.clinicaltrials.gov.
You may be interested in joining an observational trial, where there’s no treatment – only testing and examinations, typically with the goal of developing new diagnostic tests and gathering other information of potential future use in developing new treatments. If so, visit www.clinicaltrials.gov and search on PSP and, in the “focus” column, on “observational studies.” One that I’d recommend highly is the ALLFTD study. It’s centered around frontotemporal dementia but also includes PSP. I know the people running it, and it’s a first-rate study that has already recruited several hundred people with PSP. NCT04363684
Disclosures: I am, or have been, a paid consultant for the companies that make Relyvrio, NIO-752 and FNP-223. But I have no stock in the companies or other financial interest in the success of these drugs.
I could use your input right now. (Actually, I could always use your input, but only occasionally do I ever specifically ask for it.)
A few days ago, I attended a two-day conference in Washington, DC on the tau protein sponsored jointly by the Alzheimer’s Association, The Rainwater Foundation and CurePSP. A talk on clinical trials in the non-Alzheimer tau disorders mentioned the well-known difficulties in recruiting adequate numbers of patients with rare conditions like PSP. In the Q/A, I asked if there’s a realistic possibility of reducing the number of patients required for a trial by using a new approach called “personalized endpoints.” Afterwards, the editor of a journal introduced himself and asked me to write a review article/opinion piece on that issue. I said OK, but now I could use your help.
Here’s the background to my question at the conference, though many of you already know what’s in the next two paragraphs:
The typical Phase II or III clinical trial divides the patients into active treatment and placebo groups. Trials of chronic, progressive disorders like PSP measure the signs and symptoms for each patient at “baseline,” i.e., the first visit after the screening visit, using a battery of scales and tests. One of those, which for PSP, almost always the PSP Rating Scale, is deemed the “primary outcome measure.” Other measures of the drug’s effect are called “secondary” outcomes and still others under evaluation for future use are called “exploratory” outcomes.
At the end of the double-blind period, typically one year for PSP, the battery of outcome measures is repeated, many of them having been repeated at interim visits as well. Then, for each treatment group, disease progression is measured as the rate of progression (i.e., PSPRS points per year) from baseline to endpoint. If that difference is less, on average, for the active group than for the placebo group, and is large enough that the likelihood of having occurred by chance is less than 5 percent, then the result is deemed “statistically significant.” If that result is reinforced by similar results in at least some of the secondary outcome measures and if the side effects are justified by the efficacy given the disorder’s severity and availability of other treatments, the drug will then be considered for approval by government regulators.
So what’s the problem?
First of all, “statistically significant” is not the same as “clinically significant.” That means that a result too small to make a difference to the individual patient can, because of a study’s large size, reach statistical significance. The FDA knows this, of course, and relies on secondary outcome measures to verify clinical significance. But the secondary measures may under-perform statistically, or may measure only a single aspect of the disease such as cognition or balance, or may lie far from the patient’s lived experience, as do, for example, an MRI or a blood test.
Secondly, averaging the entire active drug group and the entire placebo group is a very coarse measure. That means that demonstrating a given treatment effect with statistical significance requires large numbers of patients and/or a longer study. Both issues mean more expense for drug companies, which means that fewer drugs will get tested, the trials will be longer, and effective drugs may appear to be ineffective (called a false-negative result or Type 2 error). None of those things is in anyone’s best interest.
So what’s the solution?
The PSP Rating Scale is far from perfect and various improvements have been published. While each improves upon the original in some way, none is more sensitive to change. The only outcome measure confirmed to be more sensitive to change than the PSPRS is the MRI-based measurement of brain atrophy, and that’s too far removed from actual symptoms and disability. So, we need new study designs that can squeeze more information out of fewer patients.
A more sensitive way to assess a drug’s benefit uses “personalized outcomes.” That’s where each patient being enrolled is assigned an expected endpoint PSP Rating Scale score based on how much they are likely to progress over the following year according to published research. Relevant baseline data includes things like age, sex, progression since onset, baseline PSPRS score and subsets thereof, certain MRI abnormalities, and levels of certain chemical markers in the spinal fluid or blood. At the end of the double-blind period, the active drug and placebo groups are compared with regard to how many patients did better than their own pre-defined expectation.
But how much better is “better”? Enter a concept called “minimal clinically important difference.” That’s exactly what it says: The smallest change in a test score that corresponds to a change that makes a difference to the patient or family. The trick is how to obtain this information in some sort of reliable, standardized way. For PSP, the only attempt to do this to my knowledge was published in 2016 by Dr. Sarah Hewer and colleagues, mostly from Alfred Hospital in Melbourne, Australia. They mined data from a completed, negative PSP trial that used a secondary outcome measure widely employed in clinical research, the “Clinical Global Impression of Change” scale (CGI-c). This simple, seven-point scale asks the study neurologist to decide if overall, compared to baseline, the patient is “very much,” “much,” or “minimally” improved; or unchanged; or “minimally, much, or very much” worse. Hewer et al, calculated the average degree of PSPRS worsening for patients rated by the CGI-c as “minimally worse” over the course of the year of the trial. (Of course, the CGI-c uses the neurologist’s opinion, not the patient’s or family’s, but hopefully, the neurologist relied on their input. I certainly always did whenever I completed a CGI-c.)
The minimal clinically important difference in the 100-point PSPRS turned out to be 5.7 points, with a 95% confidence interval (the range encompassing 95% of the patients) of 4.83–6.51. Now, 5.7 points on the PSPRS represents about six months’ decline for the average patient with PSP-Richardson’s syndrome, with most of the other subtypes declining a little more slowly.
Finally, here’s my question for you:
As the seven steps in the CGI-c may be too coarse or too fine, is a six-month decline really the “minimal” worsening that’s important to you? Or would a smaller or larger decline be the least you’d consider important? You or a helper can judge this in terms of your overall comfort, or your ability to perform daily activities, or a combination of the two. Note that I’m asking how many months’-worth of decline is important, not merely noticeable, which I assume would be less.
Please respond in the comments feature or by email: ligolbe@gmail.com. Thank you!
A bit over two years ago, my posts from December 19, 2021 and January 22, 2022 reported that the drug censavudine (TPN-101) was entering a Phase 2a clinical trial. Now it’s over and the results were encouraging. I choose that word carefully, and here’s why:
The drug company sponsor, Transposon Therapeutics, presented the results at a conference in Lisbon a couple of weeks ago and I’ve only now had the opportunity to see the numbers. The trial was small (42 patients, of whom 10 were on placebo) and brief (a bit less than 6 months for the double-blind phase), with an open-label extension of the same duration. This study design is typical of Phase 2a trials in neurodegenerative diseases, which are designed to study safety and tolerability because their small size and short duration can’t detect clinically meaningful benefits.
The company’s press release says, “Participants treated with TPN-101 for the entire 48-week trial duration showed a stabilization of their clinical symptoms as measured by the PSP Rating Scale (PSPRS) between weeks 24 and 48. In contrast, participants who had been on placebo from weeks 1 to 24 continued to show a worsening of the PSPRS between weeks 24 and 48, suggesting a delay of clinical benefit onset of at least 24 weeks after start of drug treatment, and lagging behind the early effects on biomarkers seen in weeks 1 to 24.”
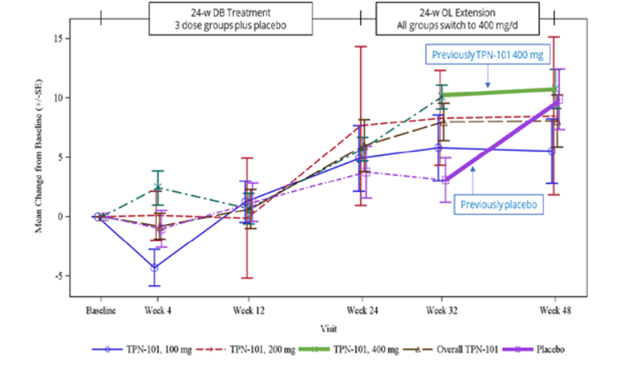
Here’s an image from the poster presented at the conference. (Sorry about the blurriness – it’s a screen shot.)
The vertical axis shows the number of points, positive or negative, by which the PSP Rating Scale changed from baseline. (Up is worse. The typical progression of PSP is 10 or 11 points per year.) The purple lines, both dashed and solid, are the patients assigned to placebo for the first 24 weeks and the other colors are the groups on the three different dosages of censavudine (100 mg, 200 mg and 400 mg per day). The brown dashed line is the aggregate of the three. The vertical line segments with hashmarks show the scores’ standard errors, a measure of variation among the patients. (See my P.S. at the end of this post.)
Note that by the end of the 24-week double-blind phase, all the results are more-or-less superimposed, meaning that the patients on active drug did no better than those on placebo. That’s normally considered a negative outcome in terms of neuroprotection, but of course this trial was too small to reveal a neuroprotective effect of realistic magnitude. After the placebo patients started receiving active censavudine at Week 24, they continued approximately along the same progressive path with the usual random wiggles (dashed and thick purple lines), ending at about 10 points worse than baseline at Week 48. This also would ordinarily be interpreted as an absence of benefit.
Now, here’s where things get tricky: After Week 32, which was eight weeks into the open-label phase, the patients on censavudine since baseline seemed to stabilize – that’s the flattening of the thick green line. In other words, their rate of progression from Week 32 to Week 48 was much less steep – a point or two rather than the expected three or four points. So, looking at the small magnitude of progression just from Week 32 to Week 48, Transposon suggested that censavudine may have a delayed benefit that became evident only after 32 weeks of treatment. In this view, the placebo group, having started censavudine at Week 24, hadn’t had enough time by Week 48 for the benefit to express itself.
Here’s the problem with that reasoning: Looking at just two visits (Weeks 32 and 48) doesn’t provide enough statistical power to conclude anything, especially with so few patients (30 patients on censavudine from the start) and only 16 weeks to work with. Furthermore, it’s easy to see that the censavudine and placebo groups, with some random wiggling, both follow the same line not only from baseline to Week 24, but also from baseline to Week 48 and from Week 24 to Week 48.
The PSPRS results were not statistically significant, of course, and Transposon didn’t claim they were. However, their presentation’s conclusions did say, “Clinical improvements emerge with longer treatment.” I say, “There’s insufficient evidence for that.”
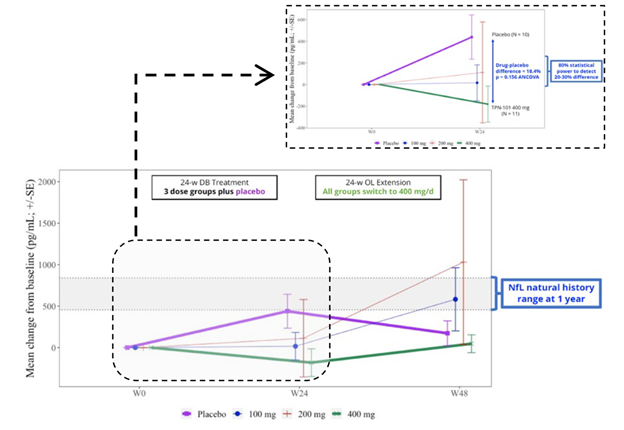
The trial included spinal fluid sampling at baseline, Week 24 and Week 48. They tested for several things, including interleukin (IL)-6, which correlates with brain inflammation, and neurofilament light chain (NfL), which correlates with loss of brain tissue in PSP. Here are the IL-6 results:
From baseline (“W0”) to Week 24, IL-6 for the group on the highest dosage of censavudine declined and continued to do so for the next 24 weeks of the same dosage. But for the groups on placebo and lower dosages of censavudine during the first 24 weeks, the IL-6 levels rose and then, after switching to censavudine (at the highest dosage), their IL-6 declined to become indistinguishable from that of the group on high-dosage censavudine from the start.
This looks very good, though I’m not sure why the placebo patients were able to “make up for lost time” so nicely. In any case, the result was not statistically significant, but at least it’s encouraging.
Now, let’s consider the NfL results, as shown in the graph below. In the group on 400 mg throughout, the level showed no change over the whole 48 weeks. By Week 24, the patients on placebo worsened to an extent predicted by previous research and then seemed to improve, or at least to stabilize, after starting to receive censavudine 400 mg. This result, like that of the IL-6, was not statistically significant, however. The 100 mg and 200 mg groups, upon switching to 400 mg at Week 24, seemed to accelerate in their degenerative course, a puzzling result. In any case, none of these observations reach statistical significance.
Despite the lack of statistical significance for the IL-6 and NfL results, Transposon’s bottom-line conclusions read, “Reductions in NfL and IL-6 support the potential for a positive effect on neuroinflammation and neurodegeneration.” Technically speaking, that nuanced statement is appropriate, as they do “support the potential,” but the very large titles for the two graphs above were the very non-nuanced “TPN-101 Reduces Neuroinflammation” and “TPN-101 Reduces Neurodegeneration.” I didn’t include them in my two screen shots because I didn’t want to perpetuate those very misleading declarations.
My bottom line: Censavudine (TPN-101) is acceptably safe over two years’ observation. This trial was too small and its double-blind phase too brief for any statement as to efficacy in slowing PSP’s progression. A large trial able to demonstrate whatever efficacy may exist is well justified and I look forward to it.
My other bottom line: Transposon’s presentation of its study’s efficacy results is misleading at best.
—
P.S. for the statistically interested: This presentation used standard errors (SE) rather than standard deviations (SD). SD is appropriate when two groups such as a placebo group and an active drug group are being compared. But for tracking the course over time of a single group, SE is appropriate. This presentation includes both kinds of observations, so I can’t fault their choice of SE. But you should keep in mind that the SE is a smaller number than the SD: SE is the SD divided by the square root of the N. So, in this trial, the N of each dosage group and the placebo group was about 10, and the square root of 10 is about 3, so the height of the error bars in each graph is only about a third of what most of us are used to looking at. That creates a false impression of a meaningful separation between the placebo and censavudine results.
A few days ago, I posted something about a clinical trial of a monoclonal antibody still accepting enrollees in Europe. I’ve since discovered that that’s incorrect, and that trial is filled. Sorry for the error. I’ve taken down that post.
But several other trials are coming down the pipeline in Europe, North America and elsewhere in the next few months. I’ll keep you informed (best I can).